

Today we are talking about medical regulation, which is the last part of our foundation. After this we will be able to assess current research and predict the future of medicine.
If you don’t know already, all medical systems, devices, and treatments are regulated. The level of oversight varies, but any technology which has direct impact on the lives of patients will be thoroughly investigated.
Regulation is very often raised as a major barrier to the use of artificial intelligence in healthcare. Today we will work out how much of a barrier it might be.
Standard disclaimer: these posts are aimed at a broad audience including layfolk, machine learning experts, doctors and others. Experts will likely feel that my treatment of their discipline is fairly superficial, but will hopefully find a lot of interesting a new ideas outside of their domains. That said, if there are any errors please let me know so I can make corrections.
Follow the leader
Today I will be talking about the US FDA, that is the American Food and Drug Administration. As much as I prefer to take an international view, it is undoubtably true that most of the regulatory bodies worldwide take cues from the FDA.
There will be some jurisdictions that adopt a more laissez faire approach (and China is certainly worth watching in this space), but by and large the US is going to lead the way.
So how does the FDA work when it comes to artificial intelligence?

We have no idea.
I’m exaggerating a little, but the truth is that the FDA has minimal guidance to assess these technologies. What this has meant in practice is that the systems have been shoehorned into old classifications that aren’t entirely relevant.
We will go over how that works in a second, but bear in mind that this is currently changing, and will change further in the future. I expect that as soon as a major breakthrough hits the market, the government is probably going to be lobbied to legislate on the issue directly.
That said, let’s see what the FDA is doing now.
A device, a drug and a doctor walked into a bar…
The FDA broadly regulates two things: drugs and devices.
The device category is broad, because it includes measuring devices, diagnostic devices, therapeutic devices and so on. It covers a really wide spectrum, from bandaids to implanted artificial hearts, data storage to artificial intelligence.
There are two main pathways through the FDA as a medical device.
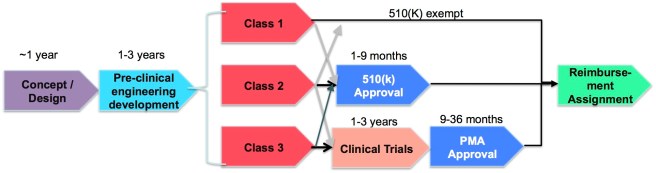
You can apply for pre-market notification, otherwise known at the 510(k) path. To get a 510(k) approval, you need to show that your device is substantially equivalent to something else already on the market. The idea is that if you device is similar enough to a device that has already been approved, then you are good to go.
The alternative is pre-market approval. This is far more rigorous, because you have to justify the device from start to finish. Safety, performance, uncertainty, usage. Essentially your device needs to be tested in real patients, and you have to show it can safely do what you claim it does.
So we can see a big problem already – no-one has done doctor-replacing AI before, so these systems cannot be substantially similar to anything on the market. In fact, “things that do doctor work” are not even usually regulated by the FDA. They are regulated by medical boards, because “things that do doctor work” are traditionally … doctors. The FDA is in uncharted territory.
This has lead some analysts to suggest:
“without a shadow of a doubt … (deep learning) products will have to be reviewed under the FDA’s premarket approval process.”
This is sort of true, but there is a third option in some circumstances.
Essentially, a device can be placed into one of three risk categories (Class I is the lowest risk, Class III is the highest). A device is automatically Class III if it is unprecedented, even if it is the equivalent of a band-aid, which means going through the rigorous pre-market approval process.
But since 2012 these sort of technologies could apply for a de novo classification. Essentially the device producer is saying “there is no similar pre-existing technology, but this doesn’t belong in Class III”.
To qualify the AI system needs to fit into Class I or II.
Class I devices are deemed to be low risk and are therefore subject to the least regulatory controls. For example, dental floss is classified as Class I device.
Class II devices are higher risk devices than Class I and require greater regulatory controls to provide reasonable assurance of the device’s safety and effectiveness. For example, condoms are classified as Class II devices.
Class III devices are generally the highest risk devices and are therefore subject to the highest level of regulatory control. Class III devices must typically be approved by FDA before they are marketed. For example, replacement heart valves are classified as Class III devices.
So, if you are considering the de novo route, the question you should ask is “is my device at least as safe as condoms?”
What this generally means is that AI systems that produce measurements can go the de novo path. A lot of effort is spent on humans (usually radiographers) identifying organs or defining the boundaries of tissues. For example, to assess the extent of coronary artery disease a human has to define which pixels are part of the coronary arteries.

I hand segmented a bunch of CT scans for some research. Every slice, every tissue. Took me months. The automated version (the “predicted” column) I put together for fun in one afternoon, and it is almost as good.
Measurements are not directly clinical, and all that matters is how accurate it is. A deep learning system like this can qualify for de novo classification, or even the 510(k) route if there is something on the market that does something similar.
We know this is true, because it has already happened. Arterys Inc obtained the first FDA approval for a deep learning system in 2016, and they got it through 510(k). Their system measures the size of the heart chambers on a cardiac MRI scan, which was a semi-automated process previously. Either the Arterys system still requires human input, or the FDA isn’t drawing a line between human-in-the-loop and human-out-of-the-loop tech, at least for measurement.
Even if it is the case that the Arterys system needs a human operator to process a case, it is definitely a nice strike against the claim that the FDA doesn’t like black box algorithms.
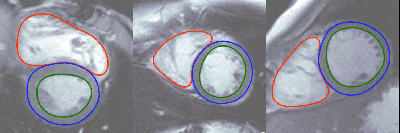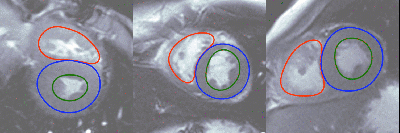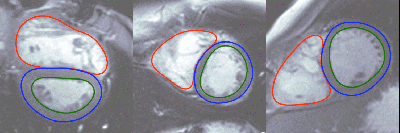
Arterys uses deep learning to do real time heart volume estimation
When describing the level of evidence the FDA required, Arterys’ CTO said:
“You need to prove statistically that your algorithm is following whatever its intended use is or [what the] marketing claims say it’s doing,”
Which is a nice, fairly low bar to pass, for measuring devices.
But measuring devices are not really what we are talking about, right?
Replacing doctors
There are two things we really want perceptual systems to do. One is highlight findings to a doctor, called CADe (computer aided detection) and the other is assessing the patient independently, called CADx (computer aided diagnosis).
The first approach has been through the FDA before, but these detection systems are generally disliked by doctors and have had poor results in practice. Highlighting possible lesions on a mammogram adds to the time it takes a doctor to review the images, and historically hasn’t achieved better outcomes.
It may be the case that these sort of systems will never work well, because they disrupt how a doctor practices. Offloading some of the process to a machine sounds fine, until we remember that doctors do most of their work subconsciously. How do you overcome decades of ingrained habits to slot a machine into the process?
Maybe it is possible if you are careful about exactly how it fits in with existing workflows, but CADx is really what we care about in this series. Replacing the doctor part entirely.
CADx has never been approved by the FDA.
CADx devices include those that are intended to provide an assessment of disease or other conditions in terms of the likelihood of the presence or absence of disease, or are intended to specify disease type (i.e., specific diagnosis or differential diagnosis), severity, stage, or intervention recommended.
This sounds like what we want deep learning to do, right?
Well, there is no guidance or rules the FDA currently applies to this group of devices. They say:
we recommend that you contact the Agency to inquire about premarket pathways, regulatory requirements, and recommendations about nonclinical and clinical data.
So, we are left to make some assumptions:
- With CADx we are replacing doctors. The tasks have a direct impact on patient care and treatment decisions. This is Class III. No question. So we need a pre-market approval.
- Clinical trials will be needed, and there are very specific recommendations on what sort of data you use. Prospective dedicated data is preferred, although retrospective and “real-world” data is sometimes allowed.
- The FDA has some wiggle room for expediting pre-market approval or de novo classification if there is “unmet medical need”. This is unlikely to be exercised unless a system is truly astonishing in safety and efficacy … which you can’t know without a phase III trial.
- You have to apply for a supplementary approval every time you want to update your systems. This is a big deal for commercial machine learning systems, where regular updates are standard practice as more data comes in.
This sounds expensive
There are some pretty massive implications in there. Returning to the earlier chart:
The pre-market approval process takes at least twice as long and even applying can cost hundreds of thousands of dollars, but the big difference here is the need for clinical trials.
Clinical trials are expensive and time consuming. We are talking years of effort and millions of dollars, most of the time.
I wrote about trials previously, and talked about the difference between phase II trials and phase III trials. So far, the only successful “doctor-replacing” medical AI studies ever performed (the Stanford dermatology paper and the Google retinopathy paper*) would be considered phase II. They were performed retrospectively, on pre-existing data obtained for other reasons, in non-clinical patient populations. Phase II studies give you some statistical evidence for how well a system works in theory.
Phase II studies don’t tell you how your system affects patients. Just because it works in the laboratory it doesn’t mean it works in clinics, and history is full of systems that worked well in phase II but failed in phase III (for this reason 50% of post phase II research never translates to clinical practice).

50% of systems that get through phase II never become products!
This is the barrier that everyone is talking about. Increased time and cost, with no promise of success.
It is worth pointing out that the clinical trials framework isn’t a perfect match for the FDA process. The FDA appears more lenient about study design and doesn’t always require randomised control trials (the hallmark of phase III studies), although the requirement for data from actual clinical cohorts seems almost universal. For a good feel for how this might affect AI, check out this paper that summarises CADe pre-market approvals in radiology (pdf link).
Talking to people in the commercial side of medical AI, the regulatory barrier is significant. Groups who would be pursuing major disruption are instead focusing their efforts on much less interesting tasks that are more likely to skip the FDA or at least have an easier run through the regulatory process. It isn’t easy to build a business model around a fifty percent chance of having a marketable product in five to ten years 🙂
The times, they are a changin’
This all depends on government priorities though, and there seems to be change in the air.

The lay of the land from one medical device consulting firm in 2016
President Trump has often expressed a desire to speed up FDA processes, as have many Republicans. But the trend is bipartisan to some extent, and last year culminated in the signing of the 21st Century Cures Act by President Obama. This bill significantly reduced the evidence requirements for some FDA processes, meaning that full clinical trials might not be required in some circumstances. While these changes applied mostly to drugs, they also touch on software.
In particular, the FDA is exempting some medical software from regulation, but these exemptions explicitly exclude any systems that take doctors out of the loop or do anything vaguely diagnostic.
The Act also allows for faster approvals in settings where there is no pre-existing technology or a humanitarian demand, but it remains to be seen how this is applied.
So it looks like the FDA is becoming less conservative, but the process is slow and so far hasn’t addressed medical AI systems. For now at least I agree with Greg Freiherr. It is hard to imagine any disruptive medical AI system that won’t need pre-market approval (and therefore serious clinical trials) to be used in the USA.
Look East
There are commercial groups trying to sidestep the regulatory barrier, with many startups looking outside of the US to other health jurisdictions. As I mentioned earlier, China has a far less restrictive regulatory framework, and there are already companies with deep learning systems being deployed in clinics. I suspect the first “real-world” data for CADx systems will come from somewhere like China.
But this series is about the displacement of doctors. China and many similar countries have severe doctor shortages, so even deployed AI might not displace doctors in these countries. The huge unmet demand could siphon up all of the productivity benefits without disrupting workforces.

Countries like China have unmet medical demand, so AI is less likely to displace doctors
So, in relation to AI disrupting medical employment, I generally agree with the received wisdom on this topic. Regulation is currently a barrier to doctor-replacing AI, and that barrier adds years of effort and millions to costs. This barrier may come down in the future, but it is likely to be a slow process. In the meantime, look to China and other similar countries for real-world results.
In fact, you could almost make a predictive rule out of that:
If you haven’t seen automation of a diagnostic medical task in China, don’t expect doctors who perform that task anywhere in the world to be displaced in the near future.
So that is all the “background reading” for this series. Next week we will get into the fun stuff – looking at the current state of the art in the published literature. How good are medical AI systems right now?
See you next week 🙂
Posts in this series
Understanding Regulation
Next post: The State of the Art

Hi Luke,
I was just reading the recently unveiled plans for the FDA to change its practices and accommodate the fast moving IT applications in medicine to allow for faster innovation (https://blogs.fda.gov/fdavoice/index.php/2017/06/fostering-medical-innovation-a-plan-for-digital-health-devices/).
Any ideas as to how this would change the points you made in this article?
And speaking about China, Infervision’s algorithm is working in several hospitals to detect lung nodules with other applications in sight.
LikeLike